DrawMol
macOS / Education
DrawMol, a powerful graphical interface to visualize and build molecular structures.
CITATION
If you use DrawMol for your scientific work, please cite the DrawMol program in your papers:
DrawMol, Vincent LIEGEOIS, UNamur, www.unamur.be/drawmol
All papers that cite DrawMol will be listed on the website
FUNCTIONALITIES
With DrawMol, you can:
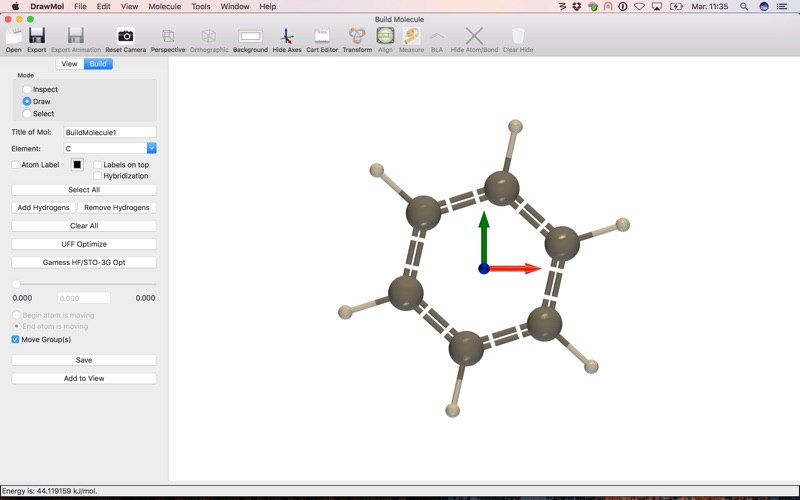
A) Build a molecular structure from scratch
- Build a structure by clicking and dragging to add atoms and bonds.
You can then refine your structure in one click using UFF force field optimization. When building a structure, DrawMol can guess for you the type of bonds (single, double, triple or aromatic bonds) or you can force it by clicking on a bond.
Moreover, hydrogen atoms can be added automatically
- Save your structure in xyz format or build a gaussian or gamess input file directly.
For Gamess, if the program is installed locally, you can even launch the job from DrawMol (in the background) and follow the progression of the job.
- Paste directly a structure into the Cartesian Editor of DrawMol. It can recognize XYZ, Gaussian, Gamess, Turbomole and Z-matrix formats.
The Cartesian Editor can also be used to output the structure in any of these formats as well as to modify a given structure by adding/removing some atoms or by changing a given atom element.
- Import a structure from ChemSpider database
B) Visualize molecular structure as well as molecular properties
- Open XYZ, MDL molfile, Gaussian log, Gaussian fchk, Gamess log, Dalton out, Molpro out file formats.
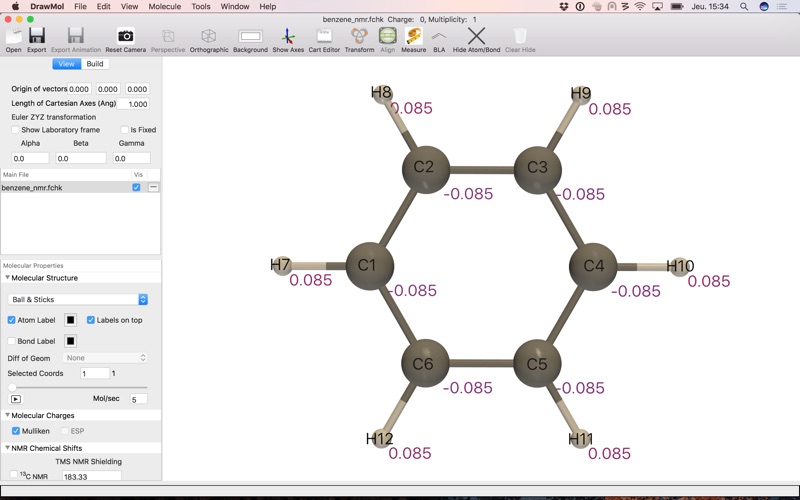
- In addition to the molecular structure, DrawMol can read various molecular properties (for now only available for Gaussian fchk file format):
. Mulliken charges
. Dipole moment
. Polarizability and first-hyperpolarizability
. Vibrational normal modes
. Magnetic shieldings (NMR)
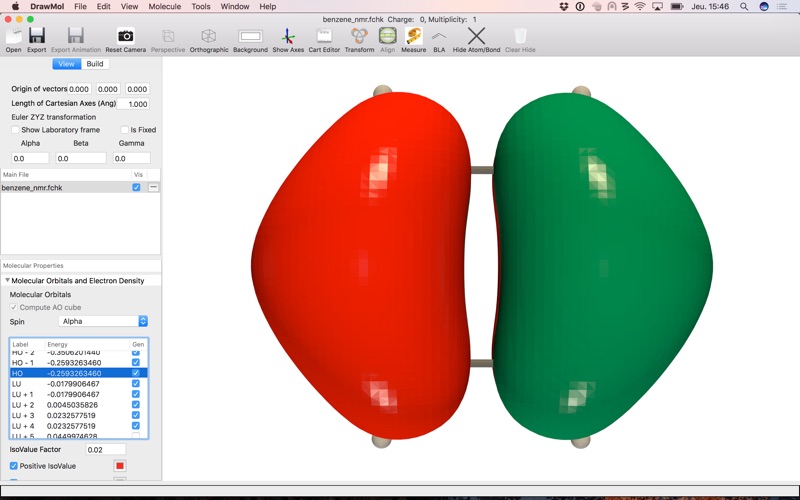
. Molecular Orbitals from the basis set and LCAO coefficients
- Visualize molecular properties:
. Atom labels
. Mulliken Charges
. NMR chemical shifts
. Dipole moment vector
. Beta vector
. Unit sphere representation of the polarizability and hyperpolarizability
. Atomic displacements of a vibrational normal modes via vectors, spheres or even animation
. Molecular Orbital isosurfaces
- Open gaussian cube files and visualize isosurfaces of the stored date.
C) In addition, DrawMol provide some tools to:
- Make two molecules or fragments to coincide with each other using a matching procedure (least-square approach)
- Orient the Z axis of a molecule to be either perpendicular to a plane made by three or more atoms or going through two or more atoms using a least-square approach.
D) Save any structure:
- As a picture either in png, tiff, jpeg or dae (Collada)
- As a movie for the animation of the normal modes
- As a XYZ, CIF, PDB files
- As a h5mol file. This format is basically a HDF5 container (https://www.hdfgroup.org) that store all the molecular properties as well as the visualization preferences.
This file can be read by DrawMol but can also be parsed easily using the C, Fortran, Python, … and even dump (with h5dump utility) into a ascii file (See https://www.hdfgroup.org for more details).
CONTACT AND HELP
If you have any issue, please send an e-mail at drawmol@unamur.be
Quoi de neuf dans la dernière version ?
- Add support for veloxchem h5 file
- Add chi1 and chi2 properties that were dropped in July 2025